Google Open-Sources ‘DeepNull’: A Method That Identifies And Adjusts For Non-Linear And Interactive Covariate Effects Using A Deep Neural Network

Each person’s genetic data contains health information that helps understand why some people are prone to diseases more than others. This answers a range of questions from why some people are at a lesser risk of developing skin cancer than others to why the effectiveness of particular treatments varies from person to person. The human genome—a DNA sequence—contains genetic information and comprises a three billion-long chain with four different nucleotides (A, C, G, and T). Only a minor portion of the genome (about 4-5 million places) differs between two people.
Genetic variants are linked to complex traits and diseases using genome-wide association studies (GWAS). Genetic studies focus on finding variations linked to certain phenotypes (for example, the risk of disorders like glaucoma). It is important that the interactions between phenotypes and principal components (PCs) of genotypes are adjusted as covariates to more correctly identify the strength of a correlation between genotype and phenotype. In GWAS, covariate correction can improve precision and eliminate confounding. According to studies, adjusting for a covariate in a linear model will enhance precision if the phenotypic distribution differs across covariate levels.
The effect of genotypes and variables on phenotype is assumed to be linear and additive in all state-of-the-art baselines. However, The assumption of linear and additive effects of covariates frequently fails to reflect underlying biology. As a result, a strategy to more completely model and compensate for phenotypic interactions in GWAS is required.
A new Google study introduces DeepNull, a new strategy for relaxing the linear assumption of covariate effects on phenotypes. DeepNull is a 5-fold cross-validation method to train a deep neural network (DNN) to predict phenotype using all covariates. The researchers constructed phenotypic predictions for all individuals after training the DeepNull model and included this prediction as an additional covariate in the association test.
DeepNull is very easy to use and requires minor changes to existing GWAS pipeline implementations. That is, only the addition of one more covariate is required to the existing GWAS pipeline to use DeepNull.
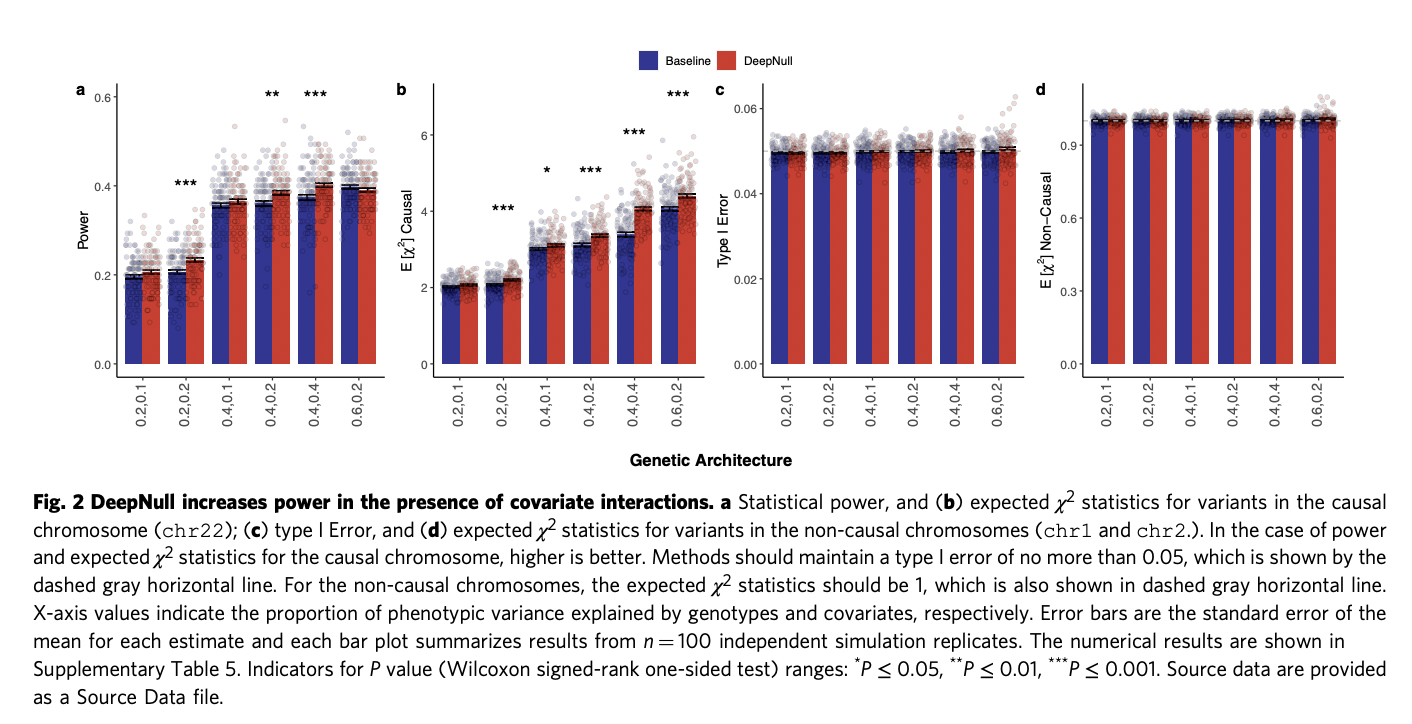
To see if DeepNull can control type I error, the researchers simulated data under various genetic topologies (genetic circumstances). They also compared DeepNull statistical power to existing state-of-the-art baselines. Initially, they simulated data in genetic architectures where variables have a linear effect on phenotype. They observed both Baseline and DeepNull could control type I error efficiently. In a scenario where covariates have just a linear effect on phenotype, DeepNull power does not diminish compared to baseline.
Further, the simulated data in genetic architectures with non-linear effects due to variables. The results show that although both baseline and DeepNull tightly control type 1 error, DeepNull boosts statistical power based on the genetic architecture. It enhances statistical power by up to 20% for specific genetic architectures.

The team used DeepNull to predict phenotypes using the polygenic risk score (PRS) and existing factors like age and gender. They looked at ten phenotypes from the UK Biobank. According to their findings, DeepNull boosts phenotypic prediction by 23% on average. They remark that DeepNull increases phenotypic prediction by 83.4 percent in the case of Glaucoma referral probability estimated from fundus pictures and by 40.3 percent in the case of LDL.
Paper: https://www.nature.com/articles/s41467-021-27930-0
Github: https://github.com/Google-Health/genomics-research/tree/main/nonlinear-covariate-gwas
Reference: https://opensource.googleblog.com/2021/12/DeepNull%20an%20open-source%20method%20to%20improve%20the%20discovery%20power%20of%20genetic%20association%20studies.html
Suggested

Credit: Source link
Comments are closed.