Researchers From Nankai and Stanford Propose ‘DeepDrug’: A Python Based Deep Learning Framework For Drug Relation Prediction

Drug discovery includes looking for biomedical connections between chemical compounds (drugs, chemicals) and protein targets. Drugs interact with biological systems on a fundamental level by binding to protein targets and influencing their downstream action. Predicting Drug-Target Interactions (DTIs) is crucial for identifying therapeutic targets or drug target characteristics. Understanding and forecasting higher-level information such as side effects, therapeutic mechanisms, and even innovative insights for drug repositioning or repurposing can all be aided by DTI knowledge.
Sildenafil, for example, was originally created to treat pulmonary hypertension, but after its adverse effects were discovered, it was repurposed to treat erectile dysfunction. Polypharmacy has also become a viable method among pharmacists because most human diseases are complex biological processes that are resistant to the activity of anyone drug. Drug-Drug Interaction (DDI) prediction and validation can sometimes identify possible synergy in drug combinations, allowing individual drugs to be more effective.
Negative DDIs, moreover, are a leading source of adverse drug reactions (ADRs), particularly among the elderly, who are more likely to take many drugs. Drugs have been withdrawn from the market as a result of critical DDIs, such as mibefradil and cerivastatin in the United States. As a result, early discovery of negative DDIs or unacceptable toxicity helps ensure medication safety while also preventing further resources from being invested in non-viable entities.
Various biochemical databases, such as DrugBank, TwoSides, RCSB Protein Data Bank, and PubChem, have emerged over the last decade, providing a quick reference for DTIs and DDIs for health professionals. Prediction of novel biochemical interactions, on the other hand, is still a difficult task. In vitro procedures are reliable, but they are both costly and time-consuming. Because of their cost-effectiveness and improving accuracy in relation to predictions, in silico techniques have gotten a lot of attention. Machine learning algorithms that combine large-scale biochemical data are used in state-of-the-art computational methods for interaction prediction.
The majority of these initiatives are founded on the idea that similar medications have similar target proteins and vice versa. As a result, the most widely used framework treats DTI and DDI prediction as a classification task using some types of similarity functions as inputs. Researchers have also looked into deep learning-based algorithms that use different feature extraction techniques in conjunction with different neural network designs, such as DeepDDI for DDI predictions and DeepDTA for DTI predictions.
Another frequent method is to use random walks to build a heterogeneous network in the chemogenomics space to forecast probable interactions. Natural language processing (NLP) techniques were used on a vast volume of relevant text corpora to automate efficient relation extraction from biomedical research articles. In the last two decades, the emergence of machine learning approaches and their integration with biomedical science has boosted drug-related research substantially.
Deep learning frameworks based on graph neural network variants such as graph convolutional networks (GCNs), graph attention networks (GATs), and gated graph neural networks (GGNNs) have recently demonstrated ground-breaking performance in social science, natural science, knowledge graphs, and a variety of other fields. GCNs have been used to solve a variety of biochemical problems, including molecular fingerprinting, where each node in the graphical model represents an atom, and each edge represents a chemical bond, and protein classification, where each node represents a residual and each edge represents the distances between nodes. Because structural qualities are the primary source of pharmacological and genetic similarities, graphical representations of biological entities have proved to be more capable of capturing structural features than Euclidean representations without the need for feature engineering.
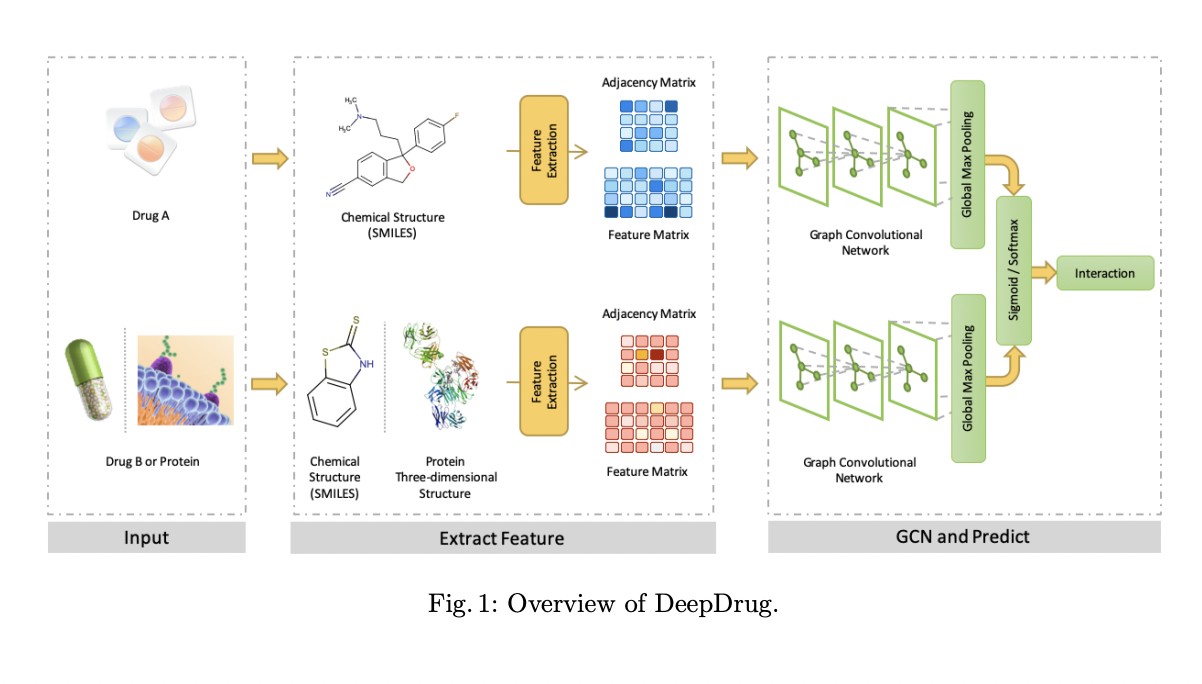
Based on these findings, Stanford University and Nankai University researchers offer DeepDrug, a graph-based deep learning framework for learning drug interactions like paired DDIs or DTIs. In the following ways, the proposed model differs from existing drug relationship prediction methods: 1) DeepDrug requires only graph representations of drugs and proteins as input to learning structural features because it takes advantage of the natural graph representation of drugs and proteins; 2) DeepDrug uses GCN modules to capture the intrinsic structure between atoms of a compound and residues of a protein.
DeepDrug can successfully learn both DDIs and DTIs from graphical features in multiple tasks such as binary classification and multi-class classification and outperforms other state-of-the-art models, according to extensive testing on several benchmark datasets. To further evaluate the model’s robustness, the team creates other datasets with varied ratios of positive and negative data. The team also shows the usefulness of the graphical model in learning structural information that is not explicitly brought into the prediction framework using visualization approaches and computing Dice similarity scores among medications in the study.
DeepDrug uses a unified framework based on GCNs to extract structural information from medicines and proteins in order to predict downstream DDIs and DTIs. In contrast to hand-crafted features (e.g., molecular fingerprints) or string-based features (e.g., SMILES sequence), the DeepDrug architecture’s innovative design can automatically capture structural information by taking into account interactions between nodes and bonds in the input graphs. DeepDrug outperforms the competition in DDI and DTI prediction tasks.
The team demonstrates the superior performance of DeepDrug through extensive experiments that include binary-class classification of DDIs, multiple-class classification of DDIs, and binary-class classification of DTIs, highlighting the strong and robust predictive power of graph presentation strategy and GCN architecture. DeepDrug’s structural properties are visualized, demonstrating the crucial discovery that biological structure can influence function, and medications with similar structures have similar targets. These findings show that DeepDrug could be a valuable tool for modeling DDIs and DTIs and thereby speed up the drug discovery process.
Because chemical medications are tiny chemicals that are often easier to turn into graphs with little ambiguity, the team first assesses DeepDrug’s performance in a binary classification context for DDI prediction. DeepDrug is compared against a baseline method based on random forest classification (RFC) and another deep learning method, DeepDDI. The baseline technique accepts graph representations, whereas DeepDDI’s initial framework only accepts SMILES strings. There are three different sets of data used.
DeepDrug regularly beats other approaches, with a 13.2 percent higher AUROC (Area Under Receiver Operating Characteristic) and a 15.1 percent higher AUPRC (Area Under Precision-Recall Curve) than the second-best method, according to the report. DeepDrug has a 31.0 percent higher AUROC and a 17.0 percent higher AUPRC than DeepDDI, owing to the fact that DeepDDI only employs SMILES sequence information as input. DeepDrug, on the other hand, makes use of a unique graph representation and has the ability to learn the underlying structural features in order to improve performance.
Conclusion
DeepDrug, a revolutionary end-to-end deep learning framework for DDI and DTI predictions, is proposed in this paper. DeepDrug takes drug SMILES strings and protein PDB inputs to characterize biological entities into graphical representations and then uses GCNs to develop latent feature representations that provide improved predictive modeling accuracy. DeepDrug can include both DDI and DTI predictions into a general framework thanks to the competitive advantage of the graph-based design. DeepDrug may now be applied to new entities with graphical representations that can be extracted.
Paper: https://www.biorxiv.org/content/10.1101/2020.11.09.375626v1.full.pdf
Github: https://github.com/wanwenzeng/deepdrug
Suggested

Credit: Source link
Comments are closed.