Google Researchers Use Machine Learning Approach To Annotate Protein Domains

Proteins play an important part in the construction and function of all living organisms. Each protein is made up of a chain of amino acid building blocks. Much like an image might have numerous things, a protein can have multiple components, known as protein domains.
Researchers have been extensively studying the challenging task of understanding the relationship between a protein’s amino acid sequence and its structure or function.
Many people are familiar with DeepMind’s AlphaFold, which uses computational methods to predict protein structure from amino acid sequences. While existing methods have successfully predicted the function of hundreds of millions of proteins, many more remain unidentified. The difficulty of reliably predicting function for highly divergent sequences is becoming increasingly serious as the volume and diversity of protein sequences in public databases grows rapidly.
The Google AI team introduces an ML technique for consistently predicting protein function. The team added about 6.8 million entries to Pfam, the widely-used protein family database that contains highly-detailed computational annotations that describe a protein domain’s function. They will be releasing it as ProtENN, which allows users to enter a sequence and receive real-time results for a projected protein function in the browser, with no setup necessary.
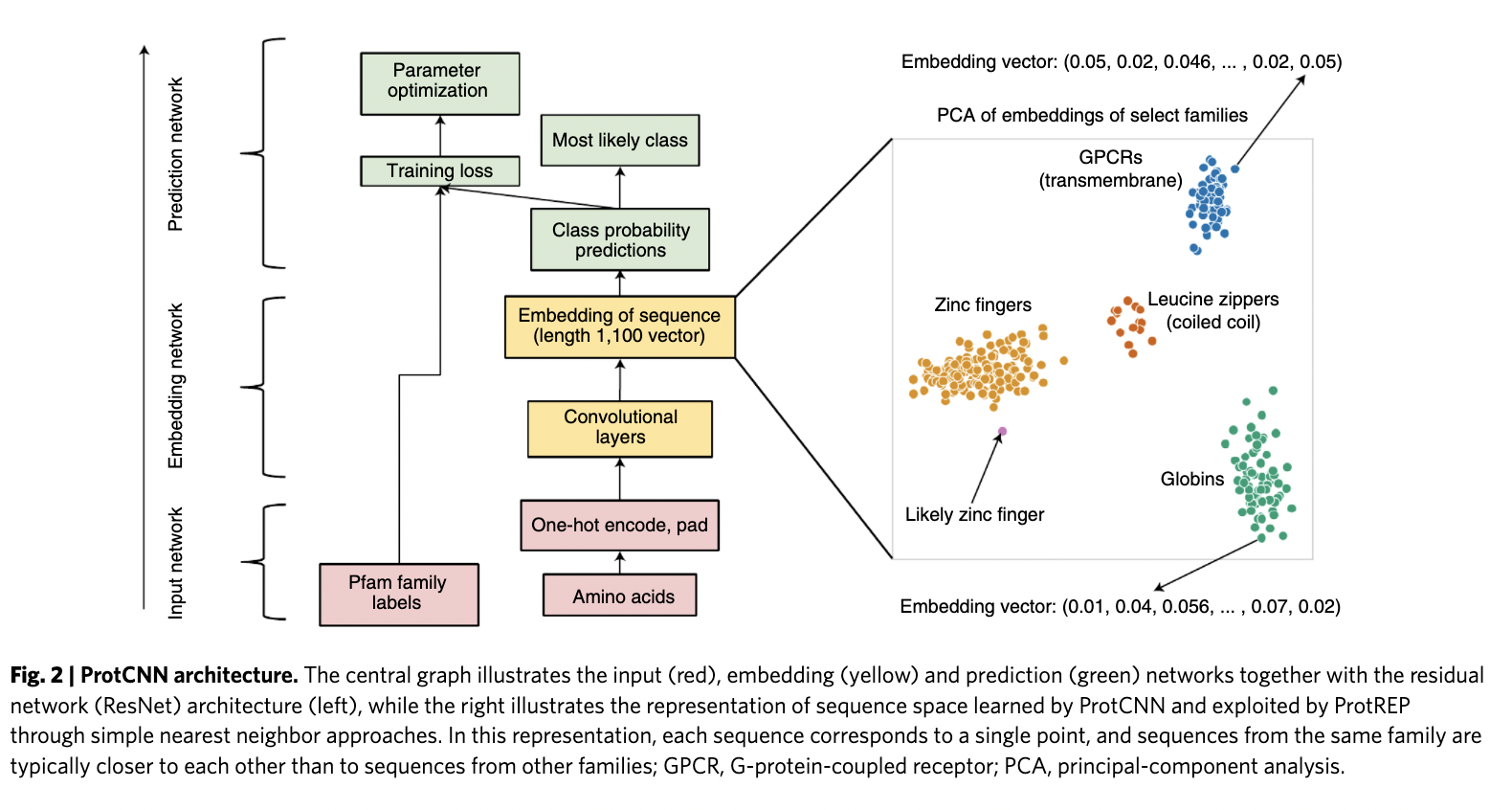
The researchers started with developing a protein domain classification model to categorize complete protein sequences. Given a protein domain’s amino acid sequence, they frame the problem as a multi-class classification task in which they predict a single label from 17,929 classes (in the Pfam database).
The major disadvantage of current state-of-the-art methods is that they are based on linear sequence alignment and do not consider interactions between amino acids in different sections of protein sequences. Proteins, on the other hand, don’t just stay as a line of amino acids. Rather, they fold in on themselves, causing nonadjacent amino acids to have strong interactions.
A fundamental stage in current state-of-the-art approaches is aligning a new query sequence to one or more sequences with established functions. Because of this reliance on sequences with known functions, predicting the function of a novel sequence that is extremely distinct to any sequence with a known function is difficult. Furthermore, alignment-based approaches are computationally costly, making them prohibitively expensive to apply to big datasets like the metagenomic database MGnify, which contains over one billion protein sequences.
The team suggests that dilated convolutional neural networks (CNNs) are well-suited to model non-local paired amino-acid interactions. In addition, they can be run on modern ML hardware such as GPUs. They train ProtCNN (1-dimensional CNNs) and ProtENN (an ensemble of independently trained ProtCNN models) to predict the classification of protein sequences.
Because proteins evolved from common ancestors, a large portion of their amino acid sequence is generally shared amongst them. It is possible that the test set is dominated by samples that are quite similar to the training data if sufficient attention is not given. This results in the models that just “memorize” the training data rather than learning to generalize it more broadly.
Therefore, it’s critical to test model performance using various setups. They stratify model accuracy as a function of the similarity between each held-out test sequence and the train set’s nearest sequence for each evaluation.
The team initially evaluates the model’s generalization ability to produce correct predictions for out-of-distribution data. For this, they used a clustered split training and test set with protein sequence samples grouped according to their sequence similarity. As whole clusters are assigned to the train or test sets, each test case differs by at least 75% from each training example.
They employ a randomly split training and test set for the second evaluation to stratify samples based on how challenging they will be to classify. The similarity between a test example and the nearest training example and the number of training examples from the genuine class are two metrics of difficulty.
They test the effectiveness of the most generally used baseline models and assessment setups, focusing on:
- BLAST, a nearest-neighbor method that employs sequence alignment to quantify distance and infer function
- Profile hidden Markov models (TPHMM and phmmer).
The team collaborated with the Pfam team at the European Molecular Biology Laboratory’s European Bioinformatics Institute (EMBL-EBI) to see if their approach could be utilized to mark real-world sequences. They combined the two approaches to identify more sequences than any method could do alone. The resulting Pfam-N, a collection of 6.8 million additional protein sequence annotations, were made available. The findings show that ProtENN learns information that is complimentary to alignment-based methods.
They examined these networks to determine if the embeddings were generally effective after observing the success of these methods and classification tests. For this, they created an interactive manuscript that allows users to investigate the relationship between model predictions, embeddings, and input sequences. They discovered that comparable sequences were clustered together in embedding space.
Furthermore, because they employed a dilated CNN as their network architecture, they could use previously-developed interpretability methods like class activation mapping (CAM) and adequate input subsets (SIS) to identify the sub-sequences important for neural networks predictions. With this method, they find that their network predicts the function of a sequence by focusing on the relevant elements of the sequence.
Paper: https://www.nature.com/articles/s41587-021-01179-w.epdf
Reference: https://ai.googleblog.com/2022/03/using-deep-learning-to-annotate-protein.html
Suggested
Credit: Source link
Comments are closed.