MIT Researchers Develop EquiBind: A Geometric Deep Learning Model That Becomes The Fastest Computational Molecular Docking Models
There is no denying the importance of new treatments after experiencing one of the worst pandemics, Covid-19. Due to new diseases, medication resistance, and the growing understanding of medical issues, previously incurable disorders can now be treated thanks to drug discovery.
There are over 1000000 possible drug-like molecules, and with the existing system, it is difficult to experiment on each of these molecules. Approval procedure needed before drugs can be utilised one of the obstacles to the developing of new drugs. This typically involves a lengthy process lasting up to ten years and costs about 2.5 billion dollars. Additionally, this approach is subject to failure at any time due to unanticipated adverse effects or experimental findings that contradict the claimed therapeutic efficacy.
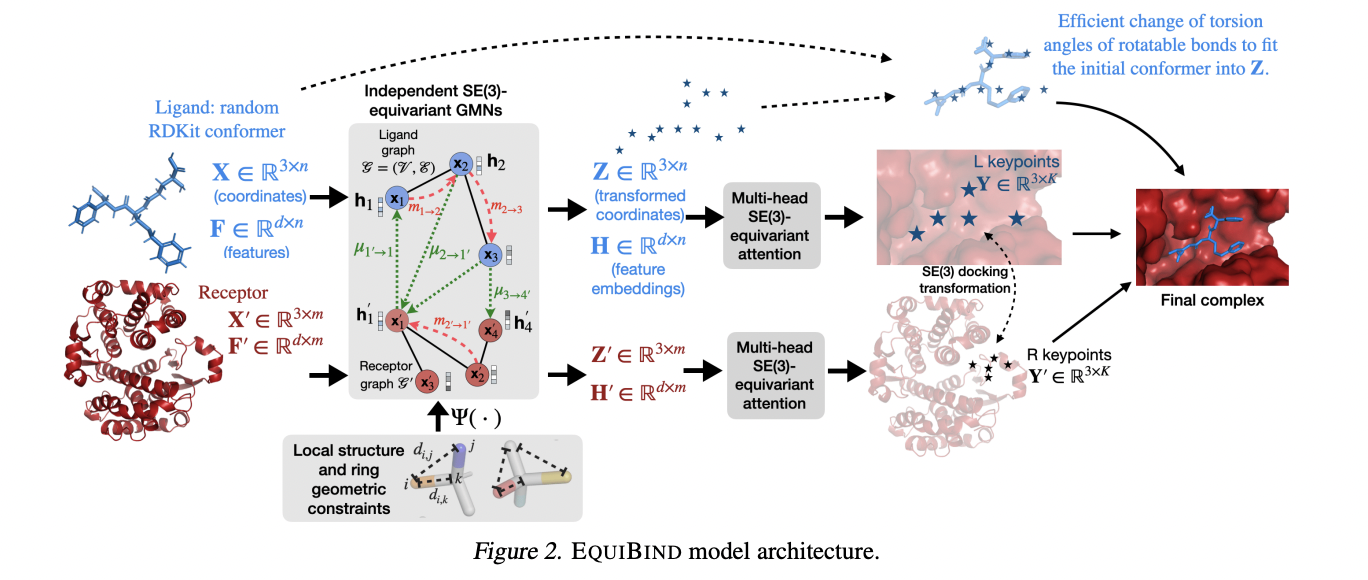
To efficiently bind drug-like compounds to proteins, MIT researchers have created a geometric deep-learning model called EquiBind that is 1,200 times quicker than one of the fastest computational molecular docking models currently in use, QuickVina2-W. EquiBind is built on its predecessor, EquiDock, which specialises in binding two proteins, as stated in their publication, “EQUIBIND: Geometric Deep Learning for Drug Binding Structure Prediction.”
Drug discovery is the process by which drug researchers identify intriguing drug-like compounds that can bind or “dock” correctly onto specific protein targets before drug development begins. The binding drug, also known as the ligand, can prevent a protein from functioning once it has effectively docked to the protein. When this occurs to a bacterium’s vital protein, the bacterium can be killed, protecting the human body.
To find the optimal “match” between the ligand and the protein, most cutting-edge computational models include substantial candidate sampling together with techniques like scoring, ranking, and fine-tuning.
According to the researchers, standard approaches to ligand-protein binding work on the trial and error method, where models spend a lot of time evaluating each “fit” before selecting the best one. EquiBind, on the other hand, uses a process called “blind docking”, which predicts the specific critical location in a single step without being aware of the protein’s target pocket.

EquiBind has built-in geometric reasoning, which helps the model learn the underlying physics of molecules and successfully generalise to make better predictions when it encounters new, unseen data, unlike most models that take multiple tries to find a favourable position for the ligand in the protein.
The group also tested their model on a protein and medicine already used to treat gastrointestinal cancers, leukaemia, and lung cancer. The results demonstrate that EquiBind successfully binded the ligands that were effective on those proteins, whereas most conventional docking techniques were unsuccessful.
EquiBind offers a special approach to the docking issue that considers pose forecasting and binding site identification. This method, which makes use of data from thousands of crystal structures that are available to the public, has the potential to change the field in unexpected ways.
Although EquiBind has gotten a lot of input from business experts that have helped the team think of real-world applications for the computational model, the team intends to uncover alternative viewpoints soon.
This Article is written as a summary article by Marktechpost Staff based on the research paper 'EQUIBIND: Geometric Deep Learning for Drug Binding Structure Prediction'. All Credit For This Research Goes To Researchers on This Project. Checkout the paper, github link and reference post. Please Don't Forget To Join Our ML Subreddit
Credit: Source link
Comments are closed.