A Novel Machine Learning Model Accelerates Decarbonization Catalyst Assessment From Months to Milliseconds
Biomass refers to organic matter, such as plants, wood, agricultural waste, and other biological materials, which can be used as a renewable energy source. It is considered a renewable energy source because it comes from living organisms and can be replenished relatively quickly, unlike fossil fuels. Biomass has the potential to be transformed into different types of energy, such as heat, electricity, and biofuels, and can potentially reduce greenhouse gas emissions and promote sustainable development.
The rural areas with farms, prairies, and ponds are a plentiful source of biomass, including corn, soybeans, sugar cane, switchgrass, and algae. These materials can be converted into liquid fuels and chemicals with a wide range of potential applications, including renewable jet fuel for all air travel in the United States.
The need for affordable and effective catalysts is a significant challenge in converting biomass into valuable products like biofuel. However, researchers at the U.S. Department of Energy’s Argonne National Laboratory have developed an AI-based model to accelerate the development of a low-cost catalyst based on molybdenum carbide.
High temperatures produce pyrolysis oil from raw biomass, resulting in a product with high oxygen content. A molybdenum carbide catalyst is employed to eliminate this oxygen content, but the catalyst’s surface attracts oxygen atoms, causing a decline in its effectiveness. To overcome this problem, researchers suggest adding a small quantity of a new element, such as nickel or zinc, to the molybdenum carbide catalyst, which reduces the bonding strength of oxygen atoms on the catalyst surface, thus preventing its degradation.
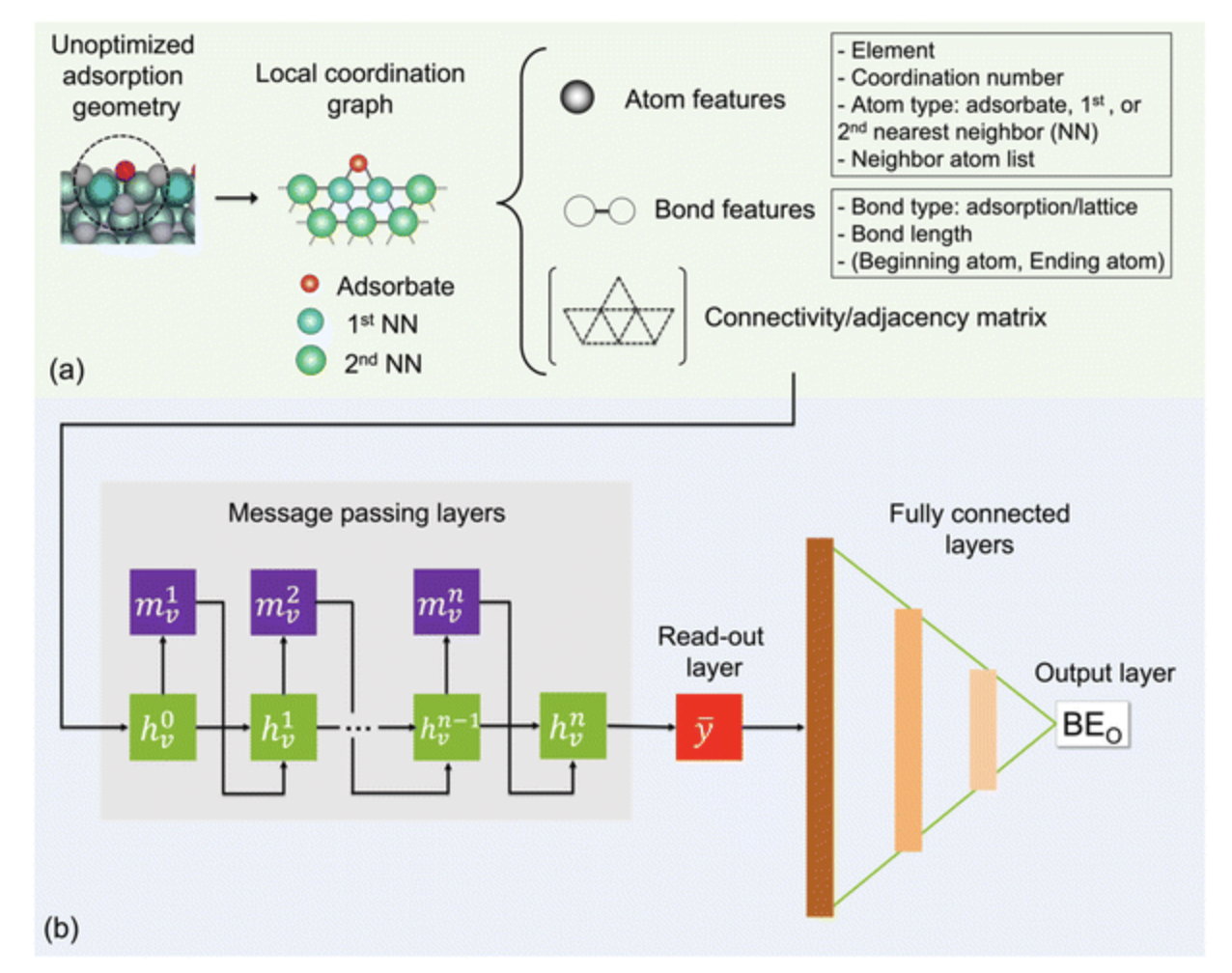
According to an assistant scientist in MSD, the challenge is to discover the best mix of dopant and surface structure to improve the molybdenum carbide catalyst’s effectiveness. Molybdenum carbide has a complex structure, so the team utilized supercomputing and theoretical calculations to simulate the behavior of surface atoms binding with oxygen and those near it.
The research team utilized the Theta supercomputer at Argonne to conduct simulations and establish a database of 20,000 structures for oxygen binding energies to doped molybdenum carbide. Their analysis considered dozens of dopant elements and over a hundred possible positions for each dopant on the catalyst surface. They then developed a deep-learning model using this database. This technique enabled them to analyze tens of thousands of structures in milliseconds, providing accurate and cost-effective results compared to conventional computational methods that take months.
The Chemical Catalysis for Bioenergy Consortium received the findings of the research team’s atomic-scale simulations and deep learning model, which they will utilize to conduct experiments and assess a shortlisted group of catalysts. According to Assary, the team hopes to expand their computational approach in the future by examining over a million structures and exploring different binding atoms, such as hydrogen. They also plan to apply the same technique to catalysts used in other decarbonization technologies, such as transforming water into clean hydrogen fuel.
Check out the paper and reference article. All Credit For This Research Goes To the Researchers on This Project. Also, don’t forget to join our 16k+ ML SubReddit, Discord Channel, and Email Newsletter, where we share the latest AI research news, cool AI projects, and more.

Niharika is a Technical consulting intern at Marktechpost. She is a third year undergraduate, currently pursuing her B.Tech from Indian Institute of Technology(IIT), Kharagpur. She is a highly enthusiastic individual with a keen interest in Machine learning, Data science and AI and an avid reader of the latest developments in these fields.
Credit: Source link
Comments are closed.