Are Pre-Trained Foundation Models the Future of Molecular Machine Learning? Introducing Unprecedented Datasets and the Graphium Machine Learning Library
The recent results of machine learning in drug discovery have been largely attributed to graph and geometric deep learning models. These techniques have proven effective in modeling atomistic interactions, molecular representation learning, 3D and 4D situations, activity and property prediction, force field creation, and molecular production. Like other deep learning techniques, they need a lot of training data to provide excellent modeling accuracy. However, most training datasets in the present literature on treatments have small sample sizes. Surprisingly, recent developments in self-supervised learning, foundation models for computer vision and natural language processing, and deep understanding have significantly increased data efficiency.
In reality, it is demonstrated that the learned inductive bias reduces the data needs for downstream tasks by spending upfront in pre-training huge models with plenty of data, a one-time expense. After these accomplishments, other research has examined the advantages of pre-training large molecular graph neural networks for low-data molecular modeling. Due to the lack of big, labeled molecular datasets, these investigations could only use self-supervised approaches like contrastive learning, autoencoders, or denoising tasks. Only a small portion of the improvement made by self-supervised models in NLP and CV has yet been produced by low-data modeling attempts by fine-tuning from these models.
Since molecules’ and their conformers’ behavior depends on their environment and is primarily controlled by quantum physics, this is partially explained by the underspecification of molecules and their conformers as graphs. For instance, it is widely known that molecules with comparable structures can exhibit significantly varying levels of bioactivity, a phenomenon known as an activity cliff, which restricts graph modeling based only on structural data. According to their argument, developing efficient base models for molecular modeling necessitates supervised training using information derived from quantum mechanical descriptions and biological environment-dependent data.
Researchers from Québec AI Institute ,Valence Labs ,Université de Montréal, ,McGill University ,Graphcore ,New Jersey Institute of Technology ,RWTH Aachen University and HEC Montré makes three contributions to molecular research. They start by presenting a brand-new family of multitask datasets that are orders of magnitude bigger than the state of the art. Second, they discuss Graphium, a graph machine learning package enabling effective training on enormous datasets. Third, various baseline models demonstrate the benefit of training on multiple tasks. They provide three comprehensive and rigorously maintained multi-label datasets, the largest currently, with approximately 100 million molecules and over 3000 activities with sparse definitions. These datasets combine labels that describe quantum and biological features that have been learned through simulation and wet lab testing, and they have been created for the supervised training of foundation models. The responsibilities covered by the labels span both the node-level and the graph-level.
The variety of labels makes it easier to acquire transfer skills effectively. It makes it possible to build fundamental models by increasing the generalizability of such models for various downstream molecular modeling activities. They meticulously vetted and added new information to the existing data to produce these extensive databases. As a result, descriptions of each molecule in their collection include information about its quantum mechanical characteristics and biological functions. The QM characteristics’ energy, electrical, and geometric components are calculated using various cutting-edge techniques, including semi-empirical techniques like PM6 and approaches based on density functional theory, such as B3LYP. As shown in Figure 1, their databases on biological activity include molecular signatures from toxicological profiling, gene expression profiling, and dose-response bioassays.

Figure 1: A visual overview of the suggested molecular dataset collections. The “mixes” are designed to be anticipated concurrently while doing several tasks. They comprise jobs at the graph level and node level, as well as quantum, chemical, and biological aspects, categorical and continuous data points.
The simultaneous modeling of quantum and biological effects promotes the capacity to characterize complicated environment-dependent features of molecules that would be impossible to obtain from what are often small experimental datasets. The Library of Graphium Has created a complete graph machine learning toolkit called Graphium to enable effective training on these enormous multitask datasets. This innovative library streamlines the creation and training of molecular graph foundation models by including feature ensembles and complicated feature interactions. Graphium addresses the limitations of previous frameworks primarily intended for sequential samples with little interaction between node, edge, and graph characteristics by considering features and representations as essential building components and adding cutting-edge GNN layers.
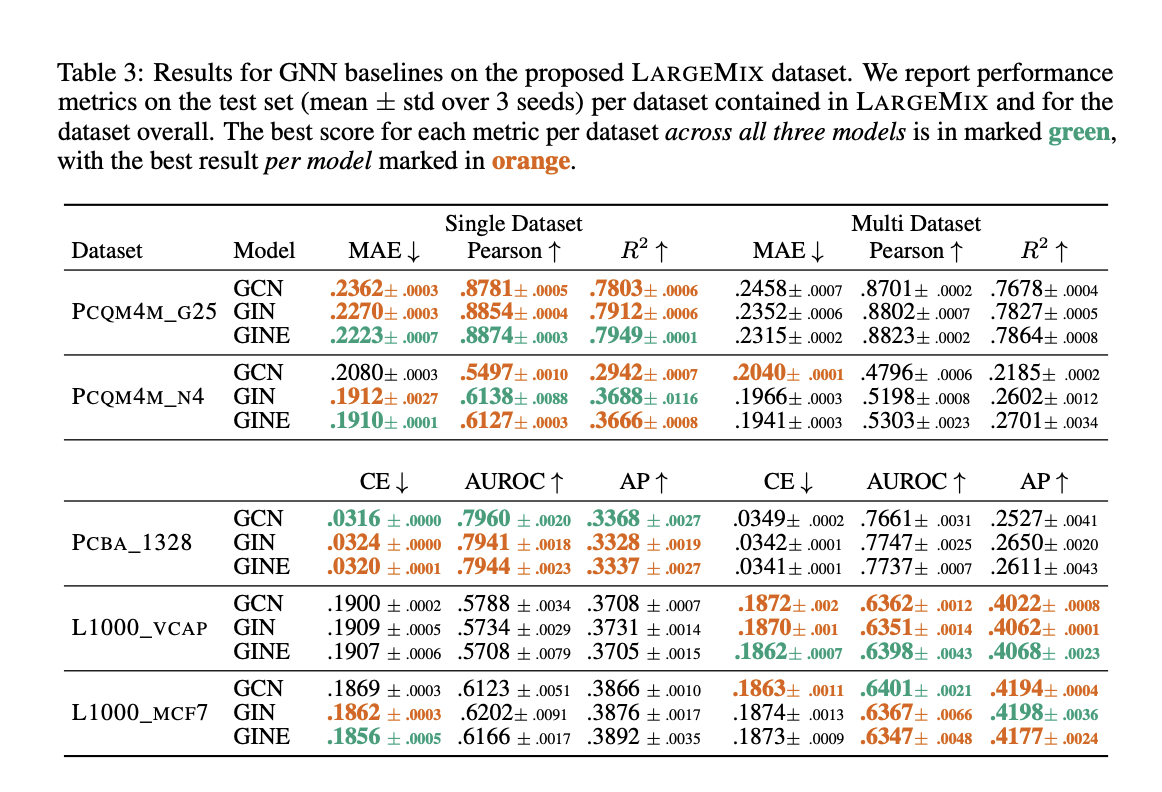
Additionally, Graphium handles the crucial and otherwise hard engineering of training models on huge dataset ensembles in a simple and highly configurable manner by offering features like dataset combination, addressing missing data, and joint training. Baseline Findings For the dataset mixtures offered, they train various models in single-dataset and multi-dataset scenarios. These provide reliable baselines that may serve as a reference point for upcoming users of these datasets and also offer some insight into the advantages of training using this multi-dataset methodology. Results for these models specifically demonstrate that training low-resource tasks may be greatly enhanced by movement in conjunction with bigger datasets.
In conclusion, this work offers the biggest 2D molecular datasets. These datasets were created expressly to train foundation models that can accurately understand molecules’ quantum characteristics and biological flexibility and, as a result, be tailored to various downstream applications. Additionally, they created the Graphium library to simplify the training of these models and provide different baseline results that demonstrate the potency of the datasets and library being used.
Check out the Paper. All Credit For This Research Goes To the Researchers on This Project. Also, don’t forget to join our 31k+ ML SubReddit, 40k+ Facebook Community, Discord Channel, and Email Newsletter, where we share the latest AI research news, cool AI projects, and more.
If you like our work, you will love our newsletter..
We are also on WhatsApp. Join our AI Channel on Whatsapp..
![]()
Aneesh Tickoo is a consulting intern at MarktechPost. He is currently pursuing his undergraduate degree in Data Science and Artificial Intelligence from the Indian Institute of Technology(IIT), Bhilai. He spends most of his time working on projects aimed at harnessing the power of machine learning. His research interest is image processing and is passionate about building solutions around it. He loves to connect with people and collaborate on interesting projects.
Credit: Source link
Comments are closed.