Northwestern Researchers have Developed a Deep Learning Approach that is Capable of Identifying the Location where a Genetic Process called Polyadenylation Occurs on the Genome
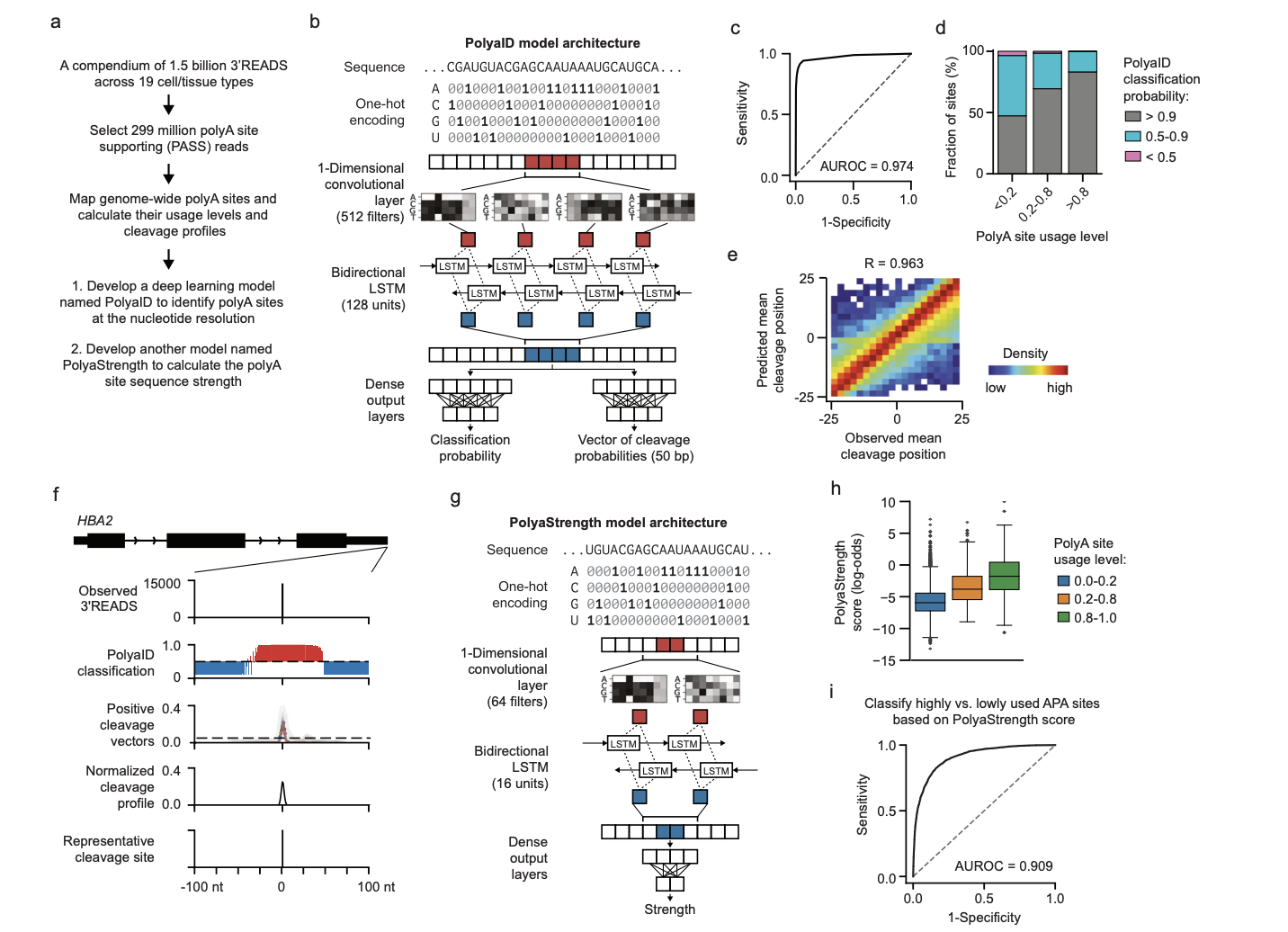
In genetics, a crucial process called cleavage and polyadenylation (polyA) ensures the proper maturation of mRNA. This process involves cutting a newly formed transcript and adding a tail of adenine nucleotides. However, if this process is not optimized with the surrounding gene structure, it can lead to premature transcription termination and the creation of abnormal proteins. Researchers from Northwestern University have developed deep learning models to understand this better across the entire human genome. These models help identify potential polyA sites with incredibly detailed precision, measuring their strength and usage in the genomic context.
Existing methods to predict polyA sites have limitations. Some models calculate the probability of a sequence being a polyA site but do not predict the exact location of the cleavage site. Others are restricted to known polyA sites, making them less versatile. The new deep learning model overcomes these challenges. It identifies potential polyA sites across the entire human genome and calculates their strength, providing a more comprehensive understanding of the process.
These models’ strength is their capacity to quantify the significance of particular motifs and their interactions during the formation of polyA sites. The polyadenylation signal (PAS) and other crucial motifs are among the distinctive cis-regulatory elements they identify, and they take into account the complex dance of different RNA-binding proteins. This means that researchers can now examine these components’ interactions and how they interact to form polyA sites in greater detail.
To demonstrate the capabilities of these models, scientists used logistic regression to study genomic parameters influencing polyA site expression in different gene regions. They found that the surrounding splicing landscape influences intronic site expression. In contrast, the usage of alternative polyA sites in terminal exons is affected by their relative locations and distances to downstream genes. This means the models identify potential sites and provide insights into how these sites are regulated based on their genomic context.
Significantly, thousands of genetic variants linked to illnesses and characteristics affecting polyadenylation activity were found using these models. This demonstrates how the models can be used practically to understand the molecular mechanisms underlying a variety of medical conditions.
To sum up, creating these deep learning models is a big step toward comprehending the intricate world of polyadenylation. Through the provision of a more refined perspective on putative polyA sites and their regulatory components, researchers can acquire a significant understanding of the molecular processes that regulate gene expression and their functions in human disorders.
Check out the Paper. All credit for this research goes to the researchers of this project. Also, don’t forget to join our 34k+ ML SubReddit, 41k+ Facebook Community, Discord Channel, and Email Newsletter, where we share the latest AI research news, cool AI projects, and more.
If you like our work, you will love our newsletter..

Niharika is a Technical consulting intern at Marktechpost. She is a third year undergraduate, currently pursuing her B.Tech from Indian Institute of Technology(IIT), Kharagpur. She is a highly enthusiastic individual with a keen interest in Machine learning, Data science and AI and an avid reader of the latest developments in these fields.
Credit: Source link
Comments are closed.