Researchers from the University of Washington Developed a Deep Learning Method for Protein Sequence Design that Explicitly Models the Full Non-Protein Atomic Context
A team of researchers from the University of Washington has collaborated to address the challenges in the protein sequence design method by using a deep learning-based protein sequence design method, LigandMPNN. The model targets enzymes and small molecule binder and sensor designs. Existing physically based approaches like Rosetta and deep learning-based models like ProteinMPNN are unable to model non-protein atoms and molecules explicitly, which limitation hinders the accurate design of protein sequences that interact with small molecules, nucleotides, and metals.
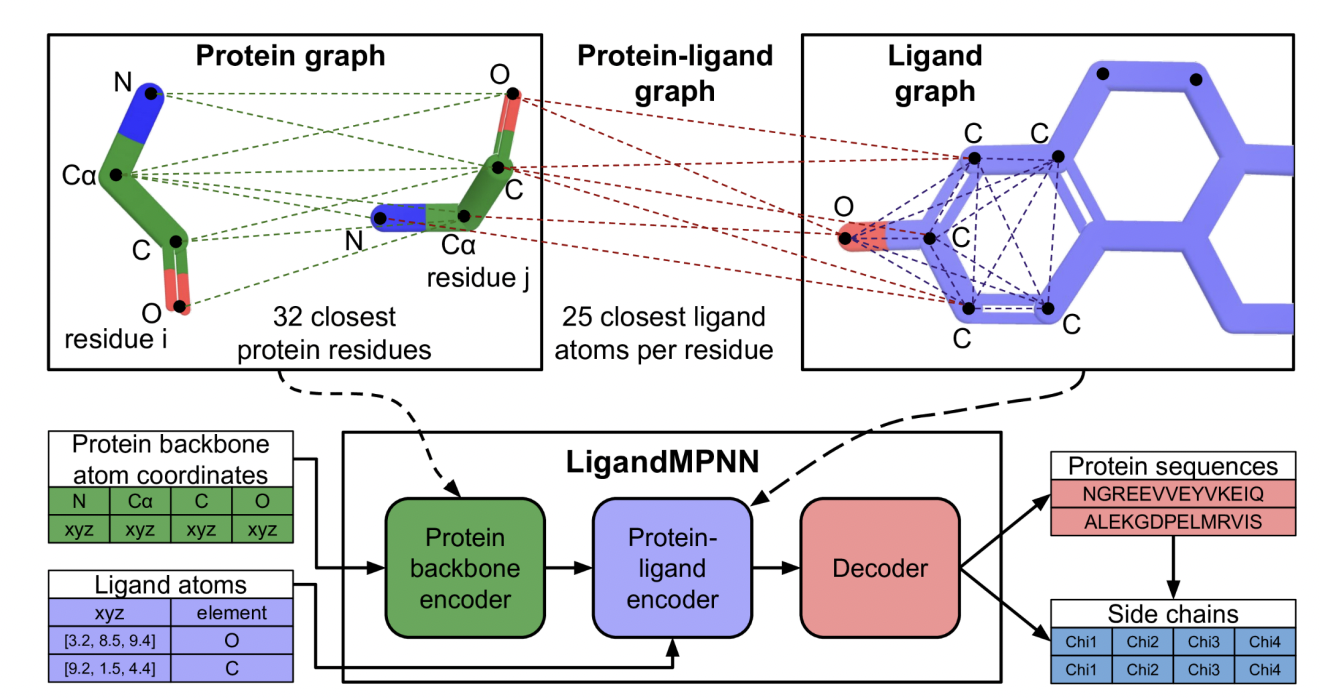
The mentioned methods neglect the explicit consideration of non-protein atoms and molecules, which is crucial for the design of enzymes, protein-DNA/RNA interactions, and protein-small molecule and protein-metal binders. The proposed solution, LigandMPNN, builds upon the ProteinMPNN architecture but explicitly incorporates the full non-protein atomic context. LigandMPNN introduces protein-ligand graphs, leveraging neural networks to model interactions and encode ligand atom geometries. The modification leads to LigandMPNN to generate sequences and side-chain conformations tailored to specific non-protein contexts.
LigandMPNN employs a graph-based approach, treating protein residues as nodes and incorporating nearest neighbor edges based on Cα-Cα distances. The model introduces protein-ligand graphs to capture interactions, with protein residues and ligand atoms as nodes and edges representing geometric relationships. The ligand graph enhances information transfer to the protein through ligand-protein edges.
The experiment demonstrated LigandMPNN and its side-chain packing better performance compared to Rosetta and ProteinMPNN, with higher sequence recovery for residues interacting with small molecules, nucleotides, and metals with 20-30% more accuracy and shows its effectiveness in detailed structural design. LigandMPNN also beats the existing models in speed and efficiency. LigandMPNN is approximately 250 times faster than Rosetta.
In conclusion, LigandMPNN fills a critical gap in existing protein sequence design methods by explicitly including non-protein atoms and molecules. The graph-based approach of LigandMPNN showcases a noticeable improvement in the performance, leading to higher sequence recovery and superior side-chain packing accuracy around small molecules, nucleotides, and metals. LigandMPNN performed exceptionally in designing small molecule and DNA-binding proteins with high affinity and specificity, which would greatly aid protein engineering.
Check out the Paper. All credit for this research goes to the researchers of this project. Also, don’t forget to follow us on Twitter and Google News. Join our 36k+ ML SubReddit, 41k+ Facebook Community, Discord Channel, and LinkedIn Group.
If you like our work, you will love our newsletter..
Don’t Forget to join our Telegram Channel
![]()
Pragati Jhunjhunwala is a consulting intern at MarktechPost. She is currently pursuing her B.Tech from the Indian Institute of Technology(IIT), Kharagpur. She is a tech enthusiast and has a keen interest in the scope of software and data science applications. She is always reading about the developments in different field of AI and ML.
Credit: Source link
Comments are closed.