Microsoft AI Researchers Develop MoLeR: A Deep Learning-Based Generative Model That Enables Efficient Drug Design
This Article Is Based On The Research Paper 'LEARNING TO EXTEND MOLECULAR SCAFFOLDS WITH STRUCTURAL MOTIFS' and Microsoft Article. All Credit For This Research Goes To The Researchers Of This Paper 👏👏👏 ✍ Submit AI Related News/Story/PR Here Please Don't Forget To Join Our ML Subreddit
Healthcare systems constantly require new drugs to address unmet medical needs across diverse therapeutic areas. Pharmaceutical industries strive to deliver new drugs to the market through the complex activities of drug discovery and development. Target identification and validation, hit identification, lead creation and optimization, and finally, the identification of a candidate for further development are all part of the discovery process. Development, on the other hand, includes optimizing chemical synthesis and formulation, doing toxicity research in animals, conducting clinical trials, and finally obtaining regulatory approval. Both of these procedures take a long time and cost a lot of money.
Expert medicinal chemists are currently working to develop “hit” molecules, which are compounds that show some potential but also some unfavorable features during early screening. Chemists aim to alter the structure of hit compounds in subsequent tests to improve their biological efficacy and eliminate potential negative effects. To focus costly and time-consuming research on the most promising compounds, computational modeling approaches have been created to forecast how the molecules will fare in the lab.
To overcome these issues, a new study by the Microsoft Generative Chemistry team in collaboration with Novartis has developed a model named MoLeR. Their paper, “LEARNING TO EXTEND MOLECULAR SCAFFOLDS WITH STRUCTURAL MOTIFS, ” demonstrates how generative models based on deep learning may aid in transforming the drug discovery process and uncovering new molecules more quickly.

The researchers also believed that automatically developing compounds that better meet project requirements than existing candidate compounds will help the drug discovery process. They remark that only a few promising molecules exist in the vast and largely unexplored chemical space. Finding them necessitates a level of imagination and intuition that pre-programmed algorithms or established rules can’t capture.
The team had previously developed CGVAE, a generative model of molecules that did well on simple synthetic tasks. However, two issues were limiting the CGVAE model’s applicability in real drug discovery:
- It can’t be naturally constrained to only explore molecules containing a specific substructure (called the scaffold)
- Due to its low-level, atom-by-atom generative procedure, it struggles to reproduce key structures, such as complex ring systems.
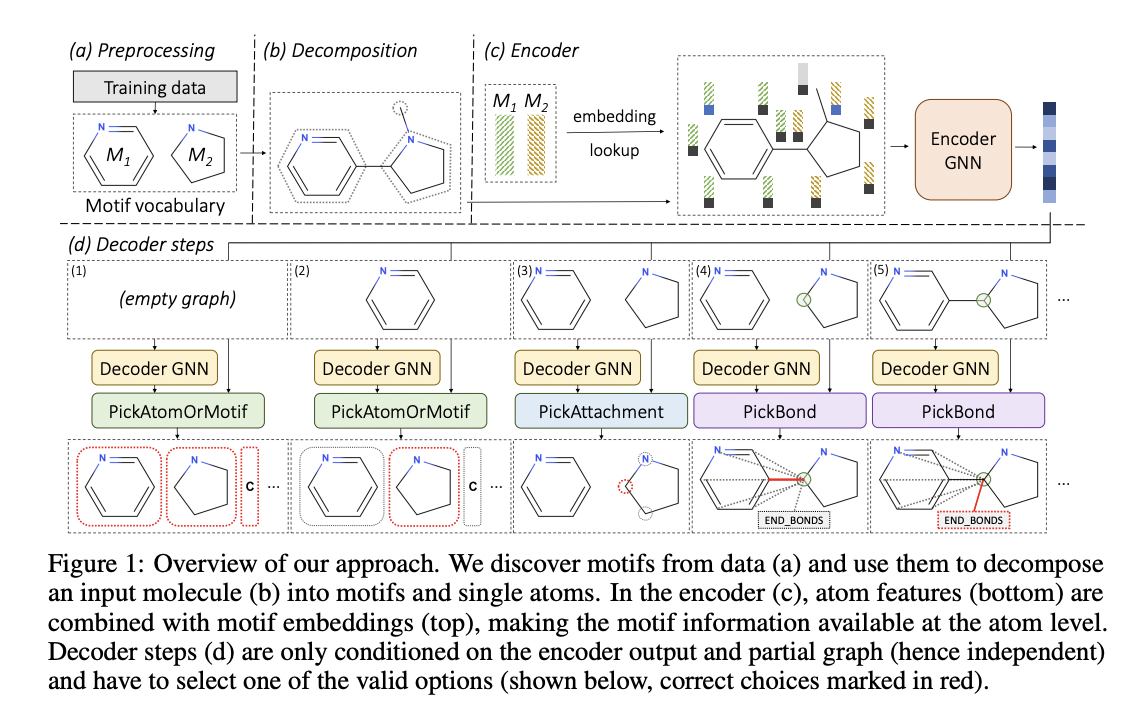
Molecules are represented as graphs in the MoLeR model, with atoms appearing as vertices connected by edges corresponding to the bonds. The team used the auto-encoder paradigm to train the model. Here, the encoder is a graph neural network (GNN) that attempts to compress an input molecule into a latent code. On the other hand, the decoder attempts to reconstruct the original molecule from this code.
The reconstruction procedure is designed to be sequential because the decoder needs to decompress a short encoding into a graph of any size. They extend the partially produced graph by adding new atoms or bonds in each phase.
Rather than relying on previous predictions, the decoder in the model makes predictions at each step simply based on a partial graph and a latent code.
The researchers explain that drug compounds are not made out of random atom combinations and are usually made up of bigger structural motifs, similar to how sentences in spoken languages are made up of words rather than random sequences of letters. Unlike CGVAE, MoLeR learns to extend a partial molecule utilizing full motifs after first discovering these common building components from data (rather than single atoms). MoLeR is also trained to build the same molecule in various orders because the order in which the molecule is built is arbitrary.
As a result, MoLeR not only requires fewer steps to make drug-like compounds, but it also does it in steps that are more analogous to how scientists think about molecule construction.
A scaffold is defined as a crucial component of the molecule that has already demonstrated promising properties. Drug development programs generally focus on a narrow subset of the chemical space by first identifying a scaffold and then only looking at compounds that contain the scaffold as a subgraph. By using an arbitrary scaffold as an initial state in the decoding loop, the design of MoLeR’s decoder allows the smooth integration of an arbitrary scaffold. MoLeR learns to complete arbitrary subgraphs via randomized generation order during training, making it suited for targeted scaffold-based exploration.
The researchers mention that although MoLeR has no concept of “molecular optimization,” it is possible to use an off-the-shelf black-box optimization method to do optimization in the space of latent codes. They used Molecular Swarm Optimization (MSO) in this work because it produces state-of-the-art results for latent space optimization in other models. Their findings show that it works well for MoLeR as well. The team tested optimization with MSO and MoLeR on new benchmark tasks that are akin to true drug discovery projects involving huge scaffolds and discovered that this combination outperformed current models.
Source: https://www.microsoft.com/en-us/research/blog/moler-creating-a-path-to-more-efficient-drug-design/
Paper: https://openreview.net/pdf?id=ZTsoE8G3GG
Github: https://github.com/microsoft/molecule-generation
Credit: Source link
Comments are closed.