MIT Researchers Propose ‘GeoMol’: A Deep Learning Model That Predicts The 3D Shapes Of Drug-Like Molecules

Scientists look for drug-like compounds that can connect to disease-causing proteins and modify their functionality to search for new therapies. They must grasp a molecule’s 3D form to understand how it will connect to specific protein surfaces. However, because a single molecule can fold in hundreds of distinct ways, solving the issue experimentally is a time-consuming and costly procedure analogous to looking for a needle in a haystack.
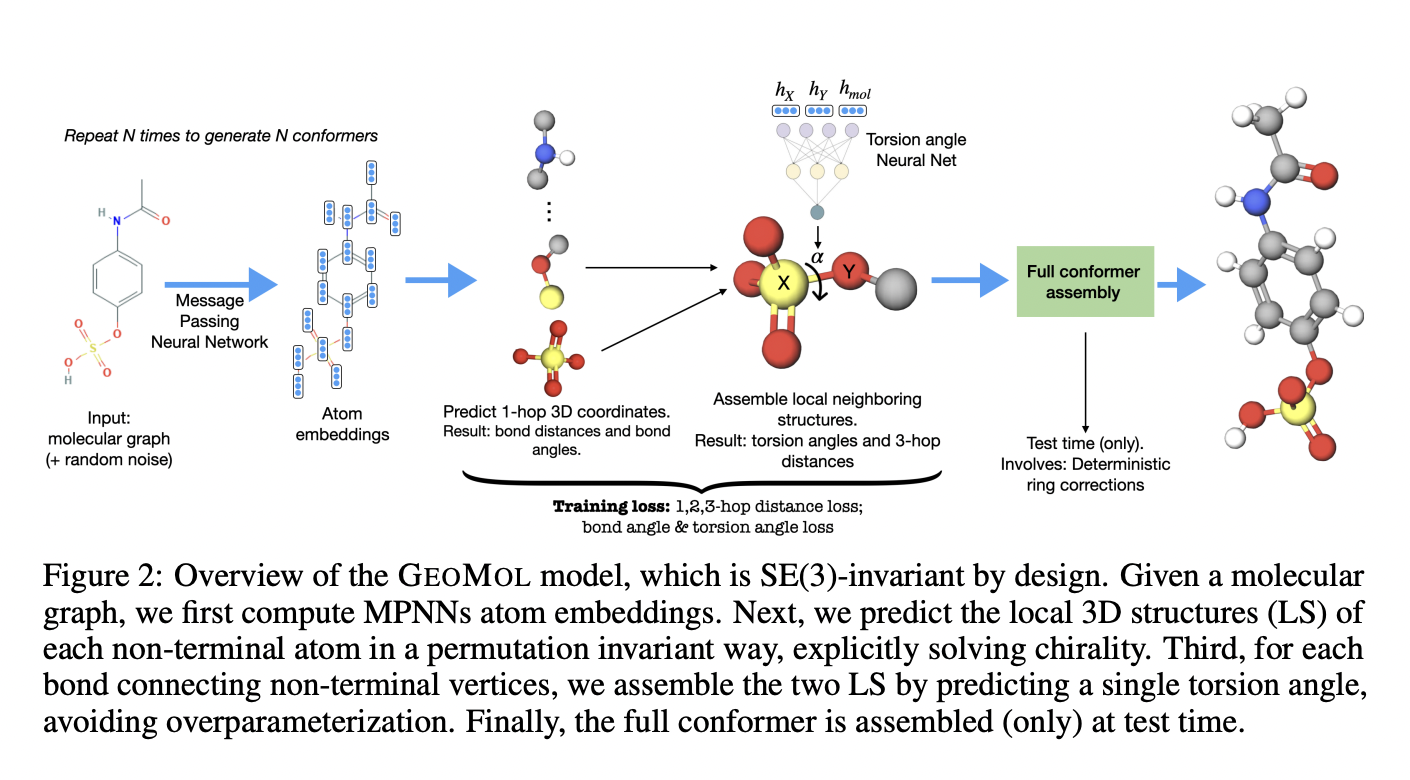
MIT researchers are using machine learning to simplify this difficult operation. They developed a deep learning algorithm that can predict a molecule’s 3D forms purely based on a 2D graph of its molecular structure. Small graphs are commonly used to represent molecules. GeoMol, their system, processes molecules in a fraction of a second and outperforms other machine learning models, including some commercial approaches. By reducing the number of compounds that need to be tested in lab tests, GeoMol could help pharmaceutical companies speed up the drug discovery process.
When we consider how these structures move in 3D space, we can see that only specific molecule sections, the rotatable bonds, are genuinely flexible. This work’s significant advance is that it approaches modeling conformational flexibility from the perspective of a chemical engineer. The model attempts to anticipate the structure’s possible distribution of rotatable bonds.
Individual atoms in a molecule are represented as nodes in a molecular graph, while chemical bonds connecting them are represented as edges. GeoMol uses a message-passing neural network; a new deep learning technology specifically intended to interact with graphs. To forecast certain components of molecule geometry, the researchers adapted a message passing neural network. GeoMol predicts the lengths and angles of chemical connections between atoms using a molecular graph. Which bonds can rotate is determined by the atoms’ arrangement and connections.
The structure of each atom’s local neighborhood is then predicted individually by GeoMol, which then assembles neighboring pairs of rotatable bonds by computing torsion angles and aligning them. The motion of three segments that are coupled, in this case, three chemical bonds that connect four atoms, is determined by a torsion angle.
The rotatable bonds can take on a wide variety of values. As a result, using message passing neural networks allows you to capture a lot of the local and global variables that impact the prediction. The rotatable bond can have many different values, and the model’s forecast can represent that underlying distribution.
Modeling chirality is one of the most difficult aspects of predicting the 3D structure of molecules. A chiral molecule, like a pair of hands, cannot be placed on its mirror image. The mirror image of a chiral molecule will not interact with the environment in the same manner. This could lead drugs to interact improperly with proteins, potentially resulting in serious adverse effects. To ensure that chirality is correctly determined, current machine learning algorithms frequently need a long, difficult optimization procedure.
GeoMol unambiguously defines chirality throughout the prediction process since it identifies the 3D structure of each bond separately, removing the need for post-process adjustment. GeoMol generates a set of plausible 3D structures for the molecule after conducting these predictions. The model can be linked end-to-end with a model that anticipates this type of protein surface attachment. The model isn’t a standalone pipeline and can easily be combined with other deep learning models.
The model was put to the test using a database of molecules and their possible 3D forms. They compared their model against machine learning models and other ways to see how many of these plausible 3D structures it could capture. GeoMol outperformed the other models on practically all of the measures that were assessed.
The model was discovered to be quite quick, which was extremely exciting to witness. Furthermore, as additional rotatable bonds are introduced, such algorithms are expected to become much slower. However, GeoMol’s performance scales nicely with the number of rotatable bonds, which bodes well for future use of these models, particularly in applications where the goal is to quickly anticipate the 3D structures inside proteins.
The researchers intend to use GeoMol in high-throughput virtual screening in the future, employing the model to find small molecule structures that interact with a certain protein. They also aim to keep adding more training data to GeoMol so that it can better anticipate the structure of lengthy molecules with many flexible bonds. Conformational analysis is an important component of increasing machine learning methodologies in drug discovery and is used in a variety of tasks in computer-aided drug design.
Paper: https://arxiv.org/pdf/2106.07802.pdf
Github: https://github.com/PattanaikL/GeoMol
Reference: https://news.mit.edu/2021/model-predicts-3d-molecule-drugs-1206
Suggested
Credit: Source link
Comments are closed.