Researchers from the University of Oxford and Xi’an Jiaotong University Introduce an Innovative Machine-Learning Model for Simulating Phase-Change Materials in Advanced Memory Technologies
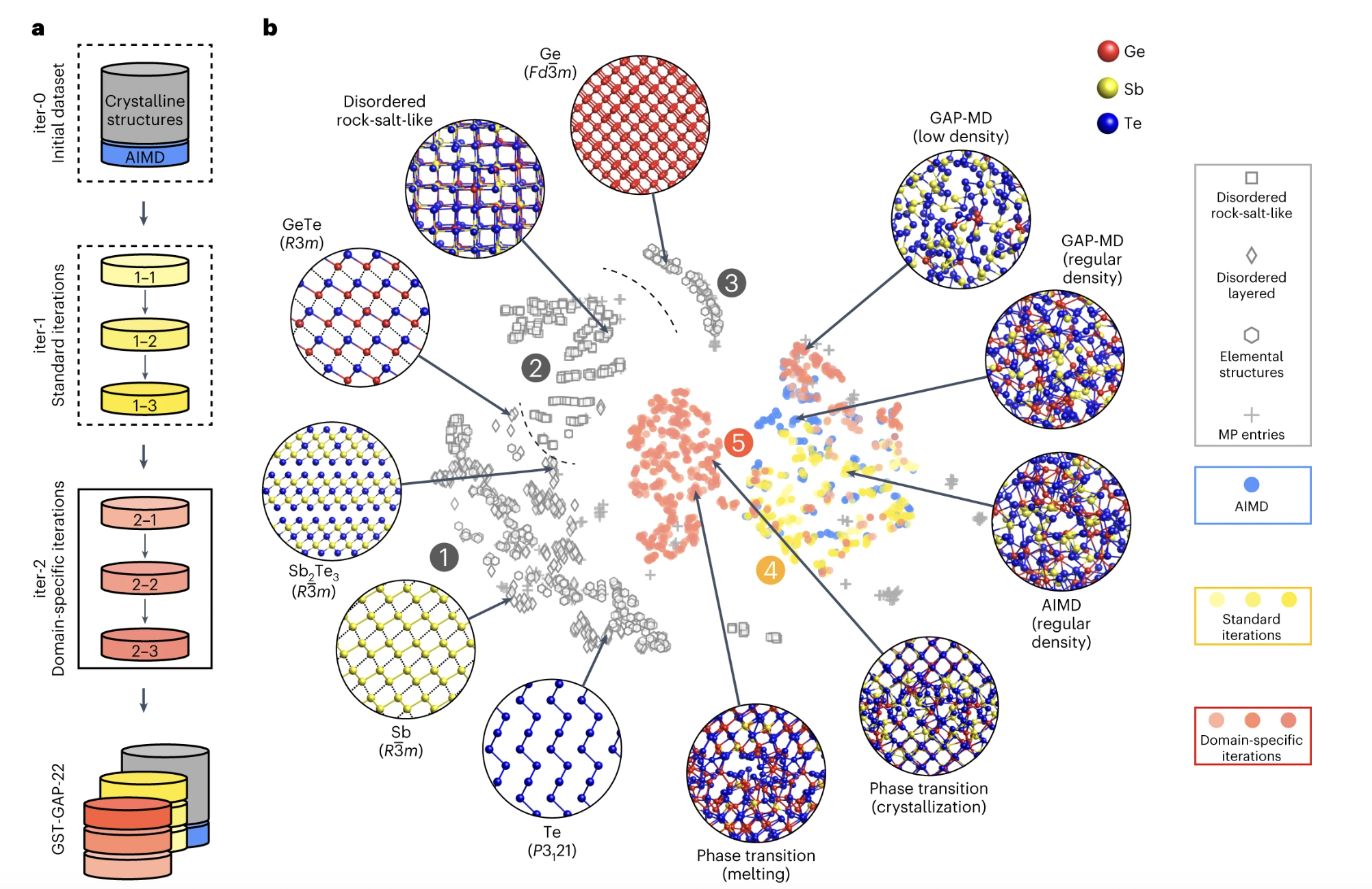
Understanding phase-change materials and creating cutting-edge memory technologies can benefit greatly from using computer simulations. However, direct quantum-mechanical simulations can only handle relatively simple models with hundreds or thousands of atoms at most. Recently, researchers at the University of Oxford and the Xi’an Jiaotong University in China developed a machine learning model that might assist with atomic-scale simulation of these materials, accurately recreating the conditions under which these devices function.
The model presented in the Nature Electronics study by the University of Oxford and Xi’an Jiaotong University can rapidly generate high-fidelity simulations, providing users with a more in-depth understanding of the operation of PCM-based devices. To simulate a variety of germanium-antimony-tellurium compositions (typical phase-change materials) under realistic device settings, they propose a machine learning-based potential model that is trained using quantum-mechanical data. Our model’s speed permits atomistic simulations of numerous heat cycles and sensitive operations for neuro-inspired computing, particularly cumulative SET and iterative RESET. Our machine learning method directly describes technologically relevant processes in phase-change material memory devices, as demonstrated by a model on the device size (40 20 20 nm3) comprising nearly half a million atoms.
Researchers demonstrate that thanks to Machine learning ML-driven modeling, fully atomistic simulations of phase shifts along the GST compositional line are possible under actual device geometries and conditions. Interatomic potentials are fitted within the GAP framework using ML for various GST stages and compositions, and the resulting reference database is then iteratively improved. The atomistic processes and mechanisms in PCMs on the ten-nanometer length scale are revealed by simulations of cumulative SET and iterative RESET processes under conditions pertinent to real operation, such as non-isothermal heating. This method enables the modeling of a cross-point memory device in a model with more than 500,000 atoms, thanks to its increased speed and precision.
The team created a fresh dataset with labeled quantum mechanical data to train their model. After constructing an initial version of the model, they gradually started feeding it data. The model developed by this group of researchers has shown great promise in preliminary tests, allowing for the precise modeling of atoms in PCMs across numerous heat cycles and as simulated devices perform delicate functions. This indicates the viability of utilizing ML for atomic-scale PCM-based device simulation.
Using a machine learning (ML) model, we significantly improved the PCM GST simulation time and accuracy, allowing for truly atomistic simulations of memory devices with realistic device shape and programming conditions. Since the ML-driven simulations scale linearly with the size of the model system, they may be easily extended to larger and more complicated device geometries and over longer timescales utilizing increasingly powerful computing resources. We anticipate that our ML model will enable the sampling of nucleation and the atomic-scale observation of the creation of grain boundaries in large models of GST in isothermal settings or with a temperature gradient, in addition to simulating melting and crystal development. As a result, the nucleation barrier and critical nucleus size for GST may be ascertainable via ML-driven simulations in conjunction with state-of-the-art sampling approaches.
Interface effects on adjacent electrodes and dielectric layers are an important topic for device engineering that could be explored in future research. For instance, it has been reported that enclosing the PCM cell with aluminum oxide walls can significantly reduce heat loss; however, the effect of these atomic-scale walls on thermal vibrations at the interface and the phase-transition capacity of PCMs cannot be studied using only finite element method simulations. It is possible to investigate this effect by employing atomistic ML models with extended reference databases to provide predictions of minimal RESET energy, crystallization time for various device geometries, and microscopic failure mechanisms to improve the design of architectures. Our results demonstrate the potential value of ML-driven simulations in creating PCM phases and PCM-based devices.
Check out the Paper. All credit for this research goes to the researchers of this project. Also, don’t forget to join our 32k+ ML SubReddit, 41k+ Facebook Community, Discord Channel, and Email Newsletter, where we share the latest AI research news, cool AI projects, and more.
If you like our work, you will love our newsletter..
We are also on Telegram and WhatsApp.
![]()
Dhanshree Shenwai is a Computer Science Engineer and has a good experience in FinTech companies covering Financial, Cards & Payments and Banking domain with keen interest in applications of AI. She is enthusiastic about exploring new technologies and advancements in today’s evolving world making everyone’s life easy.
Credit: Source link
Comments are closed.